تالاسمی
تالاسمی
تالاسمی برای اولین بار در سال 1925 توسط توماس کولی دانشمند امریکایی به عنوان یک بیماری مطرح گردید. توماس کولی پزشک متخصص کودکان متوجه کودکانی با کم خونی شدید، بزرگی طحال و تغییر شکل استخوان صورت و جمجمه شد که اغلب از خانواده های ایتالیایی و یا مهاجران از کشورهای اطراف مدیترانه بودند. وی برای نخستین بار این بیماری را تالاسمی نامید که از دو جزء تالاسا به معنای دریا و امیا به مفهوم خون تشکیل شده است و در آن سال ها به صورت بیماری کم خونی که از اطراف دریا برمی خیزد تفسیر شد.
تالاسمی به واسطه نبود یا مغایرت ژن هایی است که در ساخت هموگلوبین موثر هستند. هموگلوبین پروتئین موجود در گلبول های قرمز خون است که حامل اکسیژن هستند. افراد مبتلا به تالاسمی، نسبت به حد نرمال هموگلوبین و گلبول های قرمز کمتری تولید می کنند.
بیماری تالاسمی در دو شکل تالاسمی آلفا و تالاسمی بتا بروز می کند. بتا تالاسمی (β تالاسمی) یک گروه از اختلالات خونی ارثی ناشی از کاهش یا سنتز زنجیره بتا هموگلوبین خون می باشد. بروز سالانه در کل افراد در یک مورد در هر 100،000 در سراسر جهان تخمین زده شده است. در واقع این بیماری یکی از شایع ترین بیماری های تک ژنی در جهان است که با کاهش و یا عدم سنتز زنجیره بتاگلوبین همراه می باشد که در نتیجه آن، ساخت هموگلوبین کاهش می یابد و سبب ایجاد کم خونی می گردد.
سه شکل اصلی از این بیماری وجود دارد که شامل : تالاسمی ماژور، تالاسمی اینترمدیا و تالاسمی مینور است.
1- تالاسمی مینور یا به اصطلاح صفت تالاسمی که آنمی همولیتیک خفیف بدون علامت ایجاد می کند و در شرایط استرس زا مانند بارداری و یا عفونت های شدید، کم خونی در حد متوسط ایجاد می شود.
2- تالاسمی اینتر مدیا یا بینابینی که به صورت بزرگ شدن طحال و آنمی متوسط تا شدید بروز می کند و شکل هموزیگوت بیماری است. این بیماران قادرند هموگلوبین را بدون تزریق مکرر خون دریافت کنند. در نوع اینترمدیت بتاتالاسمی که حدود 10 درصد مبتلایان هموزیگوت را شامل می شود و شروع علایم آن از2 تا 6 سالگی است و علایم بارز قبل از تزریق خون شامل طیفی از آنمی خفیف تا علایم شدید از جمله عقب ماندگی رشد و تکامل را شامل می شود. مبتلایان اینترمدیت معمولا نیازی به تزریق خون ندارند اما به هنگام تزریق خون دچار لختگی میشوند . تالاسمی اینترمدیا یک وضعیت متوسط بین اشکال ماژور و مینور است اغلب افراد مبتلا می توانند یک زندگی عادی داشته باشند اما ممکن است تزریق گاه به گاه، مثلا در زمان بیماری و یا بارداری، بسته به شدت کم خونی خود نیاز داشته باشند.
3- تالاسمی ماژور که به آنمی کولی نیز معروف شده است و منجر به آنمی شدید می گردد و در صورت عدم تزریق خون، به نارسایی قلبی و مرگ در اوایل کودکی منجر می شود. تالاسمی ماژور طی چند ماه اول زندگی با افت سطح هموگلوبین جنینی، خود را نشان می دهد. افراد مبتلا به بتا تالاسمی ماژور معمولا در طی دو سال اول زندگی با کم خونی شدید، رشد ضعیف، و ناهنجاری های اسکلتی در طول دوره ی شیرخوارگی همراه هستند .کودکان مبتلا، به تزریق منظم و مداوم خون نیاز دارند. علایم تالاسمی ماژور از حدود 6 تا 24 ماهگی شامل مشکلات تغذیه ای، اسهال، بزرگ شدن بیش از اندازه شکم به علت بزرگی طحال و کبد، اختلال رشد، ناهنجاری های اسکلتی عمدتاً به صورت بد شکلی استخوان های بلند پا و تغییرات شکل جمجمه و عوارض مرتبط با افزایش بار آهن در قلب، کبد و غدد آندوکرینی بصورت دیابت ملیتوس و هیپوگنادیسم، هپاتیت های مزمن (هپاتیت Bیا C)، عفونت HIV، ترومبوز وریدی، مرگ به علت هموسیدروز بافت قلب می باشد.
بتا تالاسمی اینترمدیا در مقایسه با بتا تالاسمی ماژور از شدت کمتری برخوردار است و ممکن است نیاز به تزریق خون در مواقع ضروری داشته باشد.
بتا تالاسمی یکی از بیماری های خونی ارثی می باشد که از والدین به کودک منتقل می شود و بر اساس ماهیت بالینی و ارثی به دو نوع هتروزیگوت و هموزیگوت تقسیم بندی می شود. نوع هموزیگوت بتا تالاسمی بر اساس شدت اختلال ژن به دو نوع ماژور و اینترمدیت یا حد واسط تقسیم بندی می شود. نوع ماژورکه شدیدترین فرم بتاتالاسمی است نخستین بار توسط یک متخصص کودکان اهل دترویت به نام توماس کولی در سال 1925توصیف شده است.
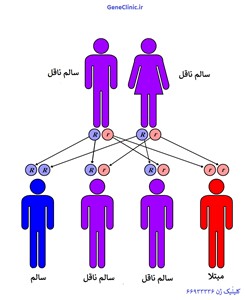
کلیه مشخصات افراد مانند رنگ چشم ؛ رنگ مو و غیره از والدین به ارث می رسد بطوری که برای هر خصوصیت دو عامل به نام ژن (عامل مولد صفات) وجود دارد که یکی از پدر و دیگری از مادر به ارث میرسد اگر شخصی تنها یک عامل (ژن) معیوب را از یکی از والدین خود دریافت کند بیمار نخواهد شد و به این شخص ناقل یا تالاسمی مینور یا مبتلا به نوع خفیف تالاسمی می گویند که می تواند عامل (ژن) تالاسمی خود را به فرزندان منتقل کند بدیهی است فرزند او نیز اگر فقط یک عامل (ژن) معیوب را از والدین خود (پدر یا مادر) دریافت کند تنها ناقل بوده و بیمار محسوب نمی شود . در حالیکه در تالاسمی ماژور یا شدید شخص دو عامل (ژن) معیوب را از والدین خود دریافت می کند (پدر و مادر هر دو ناقل هستند). اگر پدر و مادر هر دو ناقل ژن تالاسمی باشند در هر حاملگی 25% احتمال وجود دارد که کودک یک ژن معیوب را از پدر و یک ژن معیوب را از مادر به ارث برده و در نتیجه دارای دو ژن معیوب شده و مبتلا به بیماری تالاسمی ماژور گردد. ناقل تالاسمی هیچ مشکلی ندارد اهمیت ناقل بودن زمانی است که شخص توجه داشته باشد که همسر انتخابی وی ناقل نباشد .
تشخیص تالاسمی
شايعترين بيماريهاي با تظاهرات كم خوني ، كم خوني فقر آهن و بتا تالاسمي مينور ميباشند. بتا تالاسمي مينور يك بيماري تلقي نمیشود و در واقع يك مشخصه خوني است كه به ارث مي رسد . پزشكان، بتا تالاسمي مينور را با كم خوني فقر آهن اشتباه ميگيرند و درمان بيماري را بر اساس مصرف آهن قرار ميدهند. كم خوني فقرآهن هنگامي بروز ميكند كه ذخاير آهن بدن كاهش يافته و مقدار آهن موجود براي توليد طبيعي هموگلوبين كافي نباشد.
از طریق آزمایش کامل خون(CBC) و تحقیقات خاصی بر روی هموگلوبین فرد تشخیص داده میشود CBC .اطلاعات مربوط به میزان هموگلوبین و انواع مختلف گلبول های خون ، مثل گلبول های قرمز خون ، را از طریق نمونه خون در دسترس قرار می دهد . تعداد گلبول های قرمز خون و هموگلوبین در افراد مبتلا به تالاسمی کمتر از حد نرمال است . تنها نشانه در حاملان بیماری تالاسمی ، ممکن است گلبوهای قرمز آنها کمی کوچکتر از حد معمول باشند . تحقیقات هموگلوبین ، نوع هموگلوبین افراد را در نمونه ی خون می سنجد. تالاسمی به خاطر علائم و نشانه های آن از قبیل آنمی شدید ، معمولاً در اوایل کودکی تشخیص داده می شود . در برخی از افرادی که مبتلا به انواع خفیف تری از تالاسمی هستند ، پس از یک آزمایش خون معمولی ، کم خونی آنها تشخیص داده می شود . اگر کودکی مبتلا به کم خونی باشد ، پزشکان به تالاسمی مشکوک خواهند شد و او را جزء افراد در خطر ابتلا به تالاسمی قرار خواهند داد ، برای تشخیص کم خونی که از کمبود آهن ایجاد می شود و کم خونی ایجاد شده توسط تالاسمی ، آزمایش سنجش میزان آهن خون انجام می گیرد ، آنمی کمبود آهن به این علت اتفاق می افتد ، که بدن آهن کافی برای ساختن هموگلوبین ندارد اما کم خونی در تالاسمی به خاطر کمبود آهن صورت نمی گیرد و به خاطر مشکل زنجیره های گلوبین آلفا یا بتا است . مکمل های آهن هیچ تأثیری در بهبودی کم خونی ناشی از تالاسمی ندارند ، به خاطر اینکه کمبود آهن مشکل کار نیست. بررسی ژنتیکی خانواده ها برای تشخیص تالاسمی بسیار مؤثر است . در این روش سابقه ی خانوادگی مورد بررسی قرار می گیرد و هر یک از اعضاء خانواده مورد آزمایش خون و بررسی ژنتیکی قرار می گیرند.